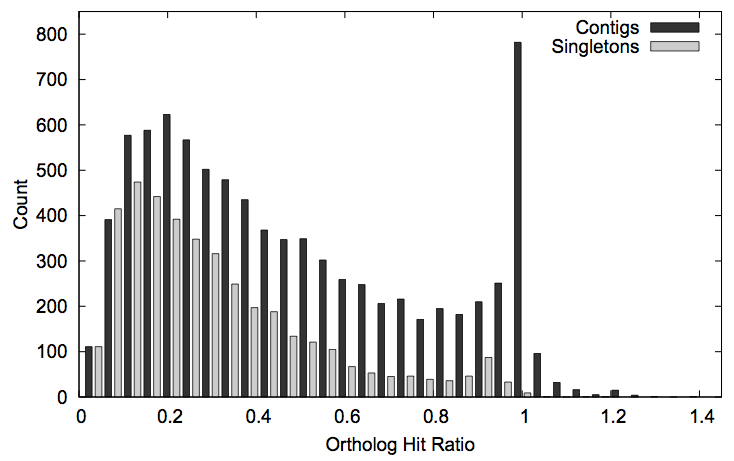
Assessing the quality and completeness of transcriptome assemblies is a challenge. For this problem, we developed a measure of gene assembly known as the “ortholog hit ratio.” First we associate each assembled gene with its closest match in a related organism. Then we compare the length of the matching region to the total length of the related gene. (Comparing the length of only the matching region ignores untranslated regions on the ends that are not considered part of the gene.) When this ratio is near 1, the sequence is likely to be completely assembled. This measure has since been adopted by other research groups. (Paper.)
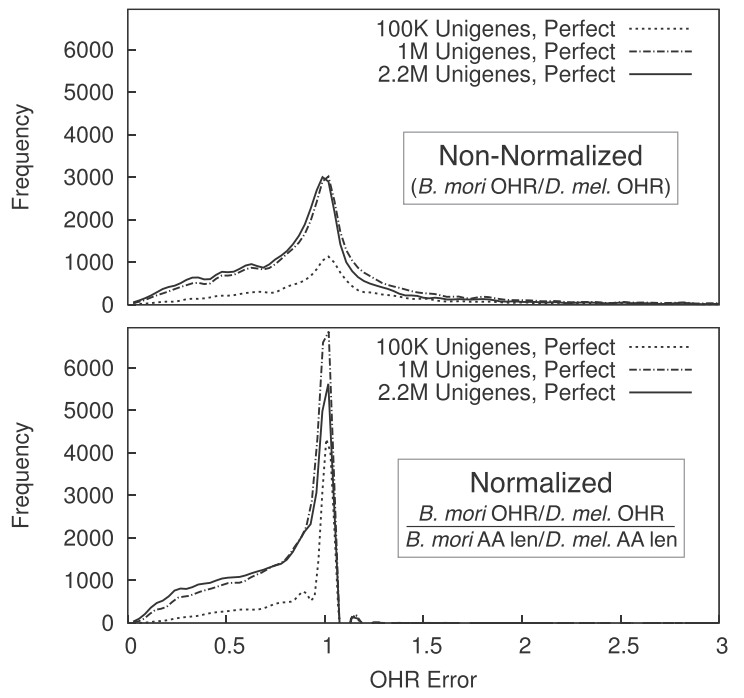
While assembly metrics for whole-genome assembly are straightfoward, those for transcriptome assembly (such as OHR above) often are not and thus it is more difficult to determine their relevance and accuracy. We simulated transcriptome data from a well-characterized species (Drosophila melanogaster) and produced both “perfect” and “realistic” assemblies, empirically testing a variety of metrics to see if they consistenty reflected the higher quality of perfect assemblies and increased dataset size. Amongst a large cache of results, we found that errors in the OHR metric are largely determined by relative expansion/contraction of transcripts in the evolutionary history of the species compared against. (Paper.)